
CAR T Breakthroughs and the Solid Tumor Challenge
Chimeric Antigen Receptor (CAR) T cells have radically improved outcomes for patients with blood cancers. With seven FDA approvals from 2017 to date for leukemia, lymphoma, and myeloma, the field has experienced unprecedented growth.
This wave of approvals has not only transformed cancer treatment paradigms but has also stimulated global investment in cell therapy platforms, manufacturing infrastructure, and the discovery and engineering of binders. Yet, the success story in hematologic malignancies has not translated to solid tumors, where CAR-T cells face a fundamentally different battleground.
CAR T cell efficacy in the solid tumor setting (e.g., lung, breast, pancreatic cancer) has been limited by various physical, molecular, and cellular factors. Unlike blood cancers, where malignant cells circulate and are relatively accessible, solid tumors present a fortress-like design. Dense stroma, immunosuppressive signaling, and antigen heterogeneity create a hostile microenvironment that actively resists cell therapy infiltration and limits its persistence.
Physical, molecular, and cellular barriers in solid tumors
| Challenge Category | Specific Challenges |
|---|---|
| Physical Barriers |
|
| Immunosuppressive Microenvironment |
|
| Antigen Heterogeneity |
|
| On-target, Off-tumor Toxicity |
|
| T Cell Exhaustion and Dysfunction |
|
| Metabolic Challenges |
|
| Limited Persistence and Expansion |
|
Major challenges associated with CAR T cell therapy efficacy in solid tumors.
In addition to the intrinsic barriers of the tumor and its microenvironment, a significant challenge to CAR T success in solid malignancies is the field’s highly experimental nature. Despite rapid global advances by researchers and developers, establishing a unified knowledge base to standardize CAR design, screening, and optimization, as well as cell processing and quality attributes of cell therapies, has been slow [1].
Lessons from blood cancer CAR T programs, such as binder tuning, vector engineering, and cell fitness metrics, must be adapted and expanded to meet the unique challenges presented by the solid tumor biology.
Why Standardized CAR Design and Functional Screening Matter
The implementation of screening approaches to objectively evaluate the performance of novel domains and their combinations is an essential step toward achieving CAR optimization and overcoming efficacy and persistence limitations in solid malignancies. Each component of the CAR molecule, including the extracellular binding domain (ECD), hinge, transmembrane domain (TMD), and intracellular domain (ICD), plays a critical role in the receptor function and must be optimized for maximal efficacy.
This complexity underscores the need for integrated screening platforms that can evaluate binder performance in the context of full CAR architecture. Functional screening in CAR format, not just binder affinity in isolation, is emerging as a critical determinant of therapeutic success.
Desirable features of CAR components for translation to solid tumors
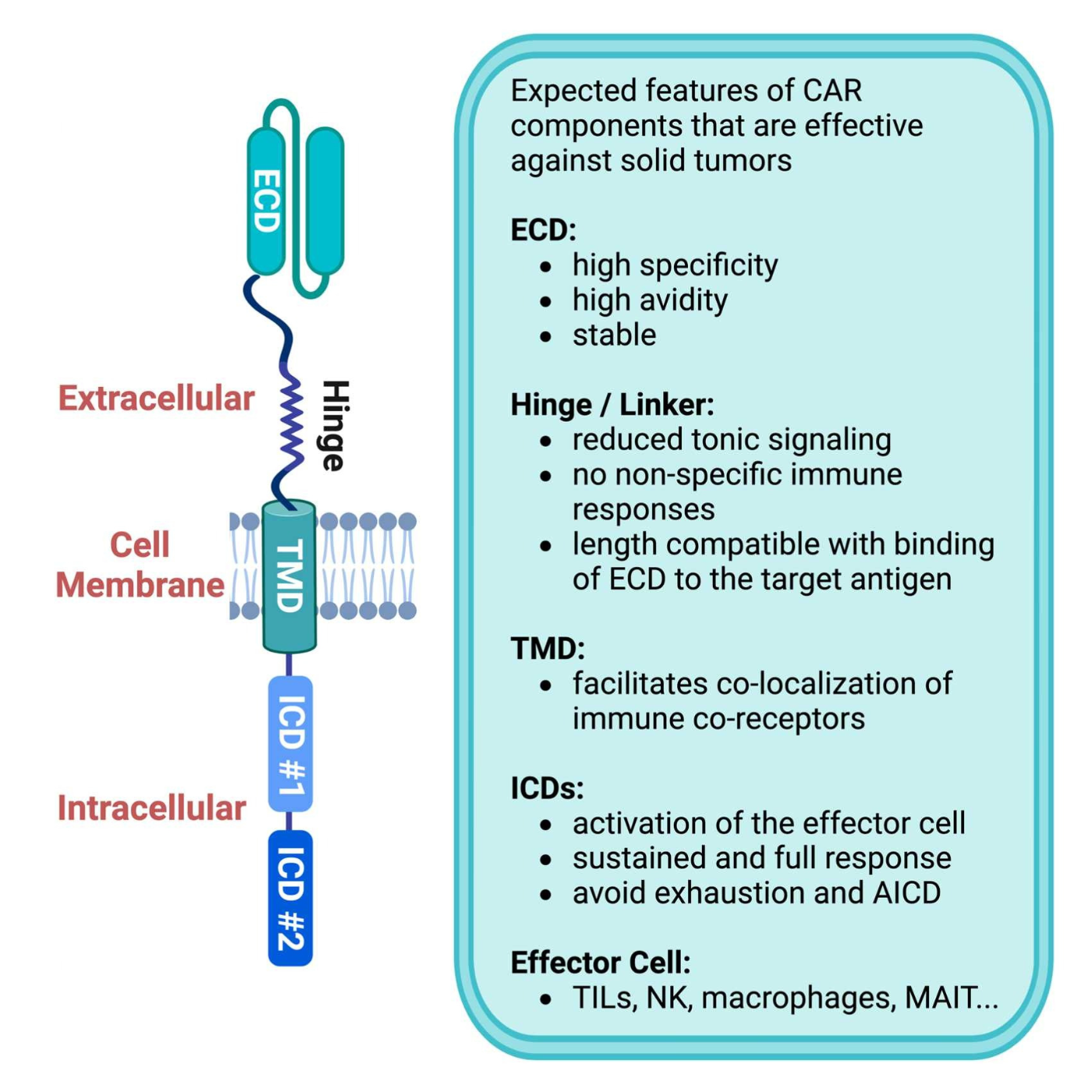
Key CAR ECD Design Considerations
The extracellular tumor antigen-binding domain enables CAR T cells to specifically interact with cancer cells and is essential for triggering receptor activation and downstream signaling. Ahead, we focus on the relevant features of CAR ECDs that require careful evaluation.
- Specificity: The specificity of the CAR binder is of utmost importance to develop cell therapies that efficiently target tumors while sparing normal tissues. This is particularly critical for solid tumors, where the majority of targets to date are overexpressed in tumor tissues but also present in healthy tissues, albeit at low levels. These tumor-associated antigens (TAAs), rather than tumor-specific antigens (TSAs), pose a significant safety risk, necessitating fine-tuning of binder affinity and avidity.
- Affinity: While high-affinity is conventionally an optimization goal for antibody-drugs, this is not necessarily a guiding principle for ECDs in CAR molecules. High-affinity CAR ECDs can lead to reduced discrimination between tumor and healthy tissues, increasing the risk of on-target, off-tumor toxicities [1]. Additionally, high-affinity ECDs have been shown to promote trogocytosis, leading to the removal of tumor-associated antigens from cancer cells and their display on the surface of CAR T cells. This process has been demonstrated in vitro for anti-CD19 CAR T cells with high-affinity binders, including the benchmark scFv FMC63, which has a documented affinity of ~2-6nM. [2,3]. Trogocytosis of TAAs by CAR T cells could adversely affect their efficacy. For example, by reducing the overall number of tumor antigens on the surface of cancer cells, trogocytosis can promote their survival. Both in vitro and in vivo animal studies have shown a connection between the use of high-affinity CARs in T cells (e.g., CD19) and reduced TAA display by cancer cells [2,4]. Moreover, the display of TAAs by CAR T cells can lead to fratricide, further diminishing cancer cell killing [4].
- Stability: The stability of the CAR ECD affects the level of tonic signaling in CAR T cells. Tonic signaling can occur through spontaneous activation of CARs, independent of tumor antigen binding, leading to the release of pro-inflammatory cytokines, such as IFN. Excessive tonic signaling can lead to cell exhaustion and reduce the efficacy of cell therapy [5].
Functional CAR Screening Streamlines Optimization
The success of CAR T cells against solid tumors depends on the careful optimization of CAR designs. The extensive fine-tuning required to optimize ECD properties necessitates robust antibody discovery platforms that provide access to TAA binder libraries of scFvs, camelid VHHs, or fully human VH domains.
Among these formats, VHH and fully human VH binders are increasingly being used in CAR development for their advantageous properties, including high stability and solubility, low immunogenicity, and greater engineering flexibility.
Increased adoption of VHH and fully human VH single domains for CAR ECDs
| ECD | Target | Indication | Stage | Date Pub |
|---|---|---|---|---|
| VHH | B7-H3 | Pancreatic cancer | Preclinical | 2023 |
| CD7 tandem | T-lymphoblastic leukemia/lymphoma | Preclinical/Clinical | 2022 | |
| CD13/TIM3 bispecific | Leukemia | Preclinical | 2020 | |
| CD19 | Lymphocytoma | Preclinical | 2023 | |
| CD20 or CD33 | Hepatobiliary carcinoma | Preclinical | 2020 | |
| CD22 | Lymphocytoma | Preclinical | 2022 | |
| CD39 | Ovarian cancer | Preclinical | 2024 | |
| CD70 | Acute myeloid leukemia | Preclinical | 2023 | |
| CD70 | Renal cell carcinoma | Preclinical | 2024 | |
| CDH17 | Neuroendocrine tumors and gastrointestinal tumors | Preclinical | 2022 | |
| CECAM6 | Pancreatic cancer | Preclinical | 2024 | |
| EGFR | Human epithelial cell carcinoma | Preclinical | 2017 | |
| FGFR4 | Hepatocellular carcinoma | Preclinical | 2024 | |
| GPC1 | Pancreatic cancer | Preclinical | 2023 | |
| GPC2 | Neuroblastoma | Preclinical | 2017 | |
| HER2 | Breast cancer | Preclinical | 2014 | |
| PDL1 | Breast and liver cancer | Preclinical | 2020 | |
| PSMA | Prostate cancer | Preclinical | 2020 | |
| Fully Human VH | CD5 | T cell leukemia or lymphoma | Preclinical | 2021 |
| CD33 | Acute myeloid leukemia | Preclinical | 2018 | |
| CD247/ALK | Solid tumors, rhabdomyosarcoma (RMS) | Preclinical | 2024 | |
| BCMA | Multiple myeloma | Preclinical | 2020 | |
| MSLN or PSMA | Acute myeloid leukemia, Prostate cancer | Preclinical | 2021 | |
| TROP2 | Solid tumors, Breast cancer | Preclinical | 2025 |
Published CAR ECDs employing VHH and Fully Human VH domains, with associated targets, indications, and development stages. Each date links to the corresponding publication source.
Once diverse libraries of binders are obtained, functional screening in the context of a CAR molecule is crucial for selecting those that support optimal efficacy and safety. While computational approaches can help discern binder stability and aggregation propensity, functional screening is ultimately more effective at guiding optimization efforts.
Screening for Antigen Discrimination
T Cell Display and CRISPR-Based Functional Screening (HER2)
To reduce cytotoxicity to healthy tissue expressing low TAA levels, an optimization strategy involves fine-tuning CAR T cell affinity/avidity. CARs engineered to achieve low TAA affinity/avidity binding in healthy tissues have demonstrated improved safety.
For example, investigators have leveraged T cell platforms to display CAR constructs, enabling in vitro screening of large libraries based on binding and activation properties. Di Roberto et al. achieved this by site-specific genomic integration of a HER2-CAR construct (i.e., into the TCR Vb chain locus) and GFP reporter (i.e., downstream of IL-2 gene) into the B3Z T cell line [6]. Next, their innovative strategy leveraged CRISPR/CAS9 editing to introduce specific mutations into the HER2-CAR ECD (i.e., anti-HER2 scFv sequence, trastuzumab). By targeting the complementarity-determining region 3 (CDRH3) of the variable heavy chain, the team generated a library of CAR binders with diverse HER2 binding properties directly in the B3Z T cell line.
Screening the resulting library through rounds of stimulation with soluble HER2 or HER2-expressing cells enabled the team to identify CAR variants with diverse binding affinities and signaling activities. Importantly, by co-culturing with cell lines with high or low HER2 surface expression (e.g., SKBR3 and MCF-7), Di Roberto et al. identified CAR ECD variants with improved antigen discrimination. Various clones were identified, with diminished binding affinity but retaining strong signaling, comparable to the original CAR, under high-HER2 expression, while showing significantly reduced signaling under low-HER2 expression.
Ultimately, screening based on signaling activation proved to be a far superior approach for identifying fine-tuned CAR ECD sequences with improved binding discrimination that couldn’t have been discovered based on HER2 binding alone.
Screening for Tonic Signaling
Tonic activity has been generally viewed as a harmful trait for CAR T cell function, leading to exhaustion phenotypes. However, recent evidence suggests that, in some instances, it can also enhance the effectiveness and longevity of specific CAR T cells (e.g., CD22 CAR T cells with a 4-1BB ICD) [5,7]. Therefore, fine-tuning tonic activity has become a relevant step in optimizing CARs to improve the performance of CAR T cells within the solid tumor microenvironment.
Most CAR T cell therapies to date have used scFv-based ECDs. Yet, the inherent instability of scFv binders has been implicated in promoting tonic signaling by inducing CAR receptor clustering and subsequent activation of CD3z signaling.8 Various strategies, such as amino acid substitutions, changes to framework regions (e.g., humanization), and adjustments to linker length in scFvs, can be leveraged to modulate binder stability and CAR tonic activity [5,8].
Quantifying Tonic Signaling (Jurkat GFP/CD69; NFAT Readouts)
Recent work by Chen et al. supports that the extent of positively charged patches (PCPs) on the surface of CAR ECDs (e.g., scFvs) correlates with the level of CAR tonic signaling [5]. To quantify CAR tonic signaling strength, Chen and colleagues engineered Jurkat cells to express CAR constructs tagged with GFP, allowing for the simultaneous tracking of CAR expression and signaling activity. They measured CD69 upregulation, an early T cell activation marker, as a functional readout, and normalized it to GFP levels to calculate a tonic signaling index for each CAR variant screened. This assay revealed that CARs, such as GD2-CAR, induced significantly higher tonic signaling than a reference CD19-CAR.
For CARs having high tonic activity, Chen et al. demonstrated that reducing PCPs on the surface of CAR binders led to reduced receptor clustering and tonic signaling [5]. The team further demonstrated that stepwise substitutions of lysine with uncharged residues in a GD2-CAR ECD progressively reduced clustering, tonic signaling, and exhaustion. In agreement with reduced exhaustion, in vivo animal studies with optimized GD2-CAR variants demonstrated improved tumor clearance and increased animal survival.
Fine-tuning charged residues within scFvs framework regions and even the CDRs in VHH domains is emerging as a necessary strategy to optimize CAR tonic activity [9]. However, modulation of tonic activity is context-dependent and varies across different CAR ECDs, requiring careful residue modification and variant screening to assess potential impacts on antigen binding properties (i.e., specificity and affinity) and antigen-dependent signaling.
Advancing CAR Optimization Through Integrated Screening
Effective CAR T development depends on integrating domain engineering with functional screening to accurately assess construct performance. This combined strategy enables systematic evaluation of CAR activity and guides optimization for solid tumor targets.
Nona Biosciences applies these principles through its NonaCAR™ and NonaCARFx™ platforms, which pair fully human VH domain binders with functional CAR-format screening for systematic construct evaluation. The system evaluates each candidate within a full CAR construct using lentiviral reporter cell lines to measure both activation and surface expression, providing a multidimensional view of binder performance. This approach helps identify binders that not only recognize the target antigen but also support stable CAR expression and signaling for improved therapeutic design.

Together, these platforms provide a modular workflow that streamlines binder discovery and construct optimization, enabling researchers to advance next-generation CAR-T, CAR-NK, and CAR-M therapies with greater precision and speed.
- Mei A. et al., Curr Opin Biotechnol, 2024. [Link]
- Hamieh M. et al., Nature, 2019. [Link]
- Seigner J. et al., Sci Rep, 2023. [Link]
- Olson M. et al., Leukemia, 2022. [Link]
- Chen J. et al., Cell Res, 2023. [Link]
- Ghorashian S. et al., Nat Med, 2019. [Link]
- Roddie C. et al., J Clin Oncol, 2021. [Link]
- Lamers C. et al., J Clin Oncol, 2006. [Link]
- Wang Y. et al., Mol Cancer, 2024. [Link]
- Singh N. et al., Nat Med, 2021. [Link]
- Landoni E. et al., Cancer Immunol Res, 2021. [Link]
- Barden M. et al., J Immunother Cancer, 2024. [Link]
- Di Roberto R. et al., Mol Ther, 2020. [Link]
- Zhou J. et al., Int Immunopharmacol, 2024. [Link]
