Mechanisms of action of less commonly used payloads
Beyond these dominant classes, several potent but less frequently used payload types contribute additional therapeutic mechanisms. DNA-damaging agents, including alkylators such as seco-DUBA, cross-linkers such as PBD dimers, and cleaving agents such as calicheamicin, offer exceptional potency but require careful control due to their broad cytotoxic potential [2]. SecoDUBA, for example, binds the minor groove of DNA and forms highly stable adducts that disrupt replication and transcription. Its membrane permeability also enables a strong bystander effect, a feature leveraged in trastuzumab duocarmazine (SYD985), now in Phase 3 evaluation [2,5,6].
Protein synthesis inhibitors represent another niche category. Payloads such as PE38 and ETA252608, derived from Pseudomonas aeruginosa exotoxin A, block peptide elongation by ADPribosylation of the eukaryotic elongation factor 2 (eEF2) [2,7]. Although rarely used, these agents demonstrate the breadth of mechanisms available to ADC developers.

Emerging ADC linkers
The push to improve linker performance has sparked a wave of innovation aimed at enhancing stability, reducing off-target cleavage, and enabling more precise control over payload release.
Improved Val-Cit linkers
Despite their success, traditional Val-Cit–PAB linkers face challenges, including aggregation, DAR constraints, and susceptibility to premature cleavage by plasma enzymes such as Ces1C and neutrophil elastase [20,21,22]. These vulnerabilities can lead to early payload release and dose-limiting toxicities. To address this, developers have incorporated hydrophilic scaffolds such as PEG or modified peptide sequences (e.g., Glu-Val-Cit, Glu-Gly-Cit) to reduce enzymatic sensitivity and improve stability [20].
More recently, repositioning the Val-Cit motif to the exo-position of the p-aminobenzyl carbamate (PAB) moiety has yielded “exo-linkers” with enhanced hydrophilicity, reduced aggregation, and improved intracellular release profiles [20].
Site-specific linker–payload conjugation
Another major frontier is the shift from heterogeneous to site-specific conjugation. Traditional lysine- or cysteine-based conjugation produces ADCs with variable DAR and inconsistent pharmacokinetics [23,24]. To overcome this, developers are leveraging engineered amino acids, introduced cysteines, or enzyme-mediated conjugation strategies.
Microbial transglutaminase (MTG), for example, enables precise conjugation at Q295 in the Fc region, though this typically requires modification of the nearby N297 glycan. A newer approach pairs MTG with a minimal RKAA peptide linker, enabling direct conjugation to native antibodies and producing homogeneous DAR2 ADCs without altering Fc glycosylation [23]. This represents a meaningful step toward more predictable and manufacturable ADCs.
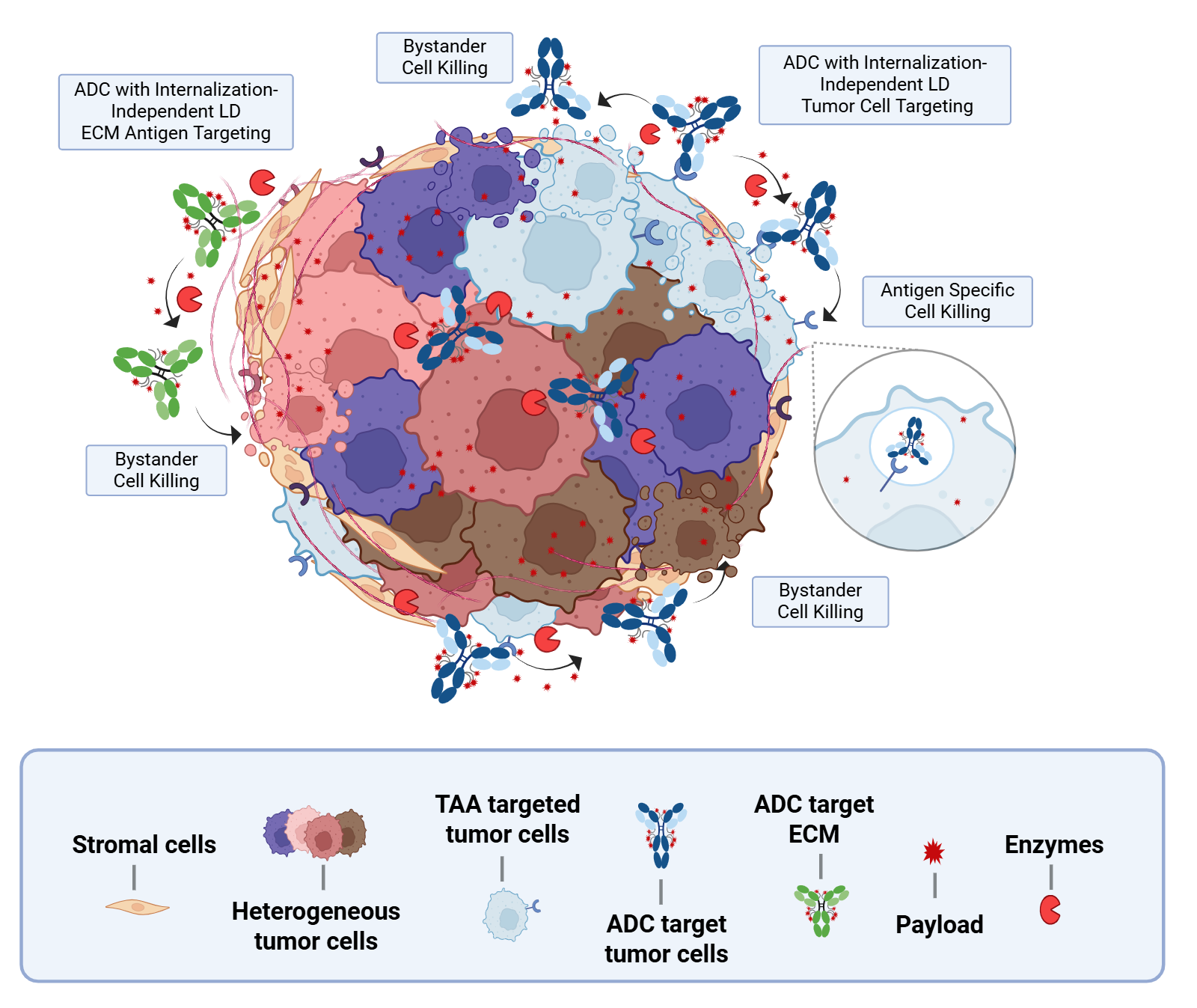
Tumor-site extracellularly cleaved linkers
A particularly exciting direction involves linkers designed to be cleaved not inside tumor cells, but within the tumor microenvironment (TME) itself. These linkers exploit enzymes enriched in the TME, such as matrix metalloproteinases, cathepsins, β-glucuronidase, and serine proteases like uPA, to trigger extracellular payload release [25,26,27].
By bypassing the need for internalization, these systems can overcome antigen heterogeneity and enhance bystander killing, broadening the therapeutic reach of ADCs. This strategy is especially promising for solid tumors, where dense stroma and variable antigen expression often limit traditional ADC efficacy.
Nona Biosciences’ proprietary TME-cleavable linker technology exemplifies this emerging class, enabling extracellular payload release driven by tumor-intrinsic enzymes. This approach is being actively advanced in discovery and preclinical programs aimed at expanding ADC utility across challenging solid tumor settings.
Innovative ADC Linker Technology for Extracellular Payload Release